COVID-19 Test Using PCR and SN&N Engineering
With the emergence of pathogens such as swine flu, Zika, and SARS-CoV-2 in the past decade, the ability to detect the presence of viruses in patient samples is critical to effective treatments and public health interventions. When novel viruses cause symptoms that overlap with those of the common cold or seasonal flu, the molecular identification of the source of infection becomes particularly vital, as evidenced by the ongoing COVID-19 pandemic.
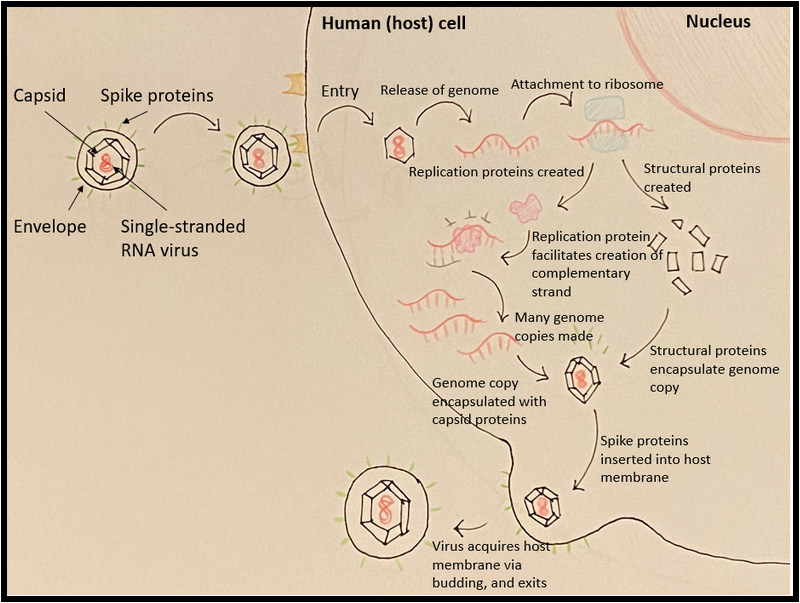
When viruses enter human cells, they hijack our intrinsic cellular machinery to replicate their own nucleic acid genomes and produce more infectious entities. Specifically, viruses in the coronavirus family have an RNA genome, encased by a protein capsule and an envelope. This envelope fuses with our cell’s membrane via specialized proteins and eventually dumps its genome into the inside of the cell, as shown in Figure 1. Our cells naturally use proteins called ribosomes to read our own RNA to make proteins vital to cellular function; with its genetic material already entering the cell in RNA form, coronaviruses can immediately take advantage of this by recruiting our ribosomes to make its own proteins. Ultimately, our cells are beaten at their own game; the tools they use to maintain themselves are stolen by the virus to replicate its genome, build its own capsule, and escape from the cell in hordes to find the next victim cell.
Blood, saliva, or other fluid samples from infected individuals can contain these viral genome copies; therefore, detecting the foreign nucleic acid product provides a definitive way for clinicians to know their patients’ infection status. Utilizing polymerase chain reaction (PCR) to amplify target DNA is a rapid, inexpensive, and sensitive way to detect the presence of a virus. The technique invented in 1985 by Kary B. Mullis, allowed scientists to make millions of copies of a scarce sample of DNA. This ground-breaking work earned Mullis the 1993 Nobel Prize in Chemistry.
A PCR reaction requires several key components: primers, polymerase, buffer, deoxyribonucleotide triphosphates (dNTPs), and the target DNA of the pathogen. Primers are short pieces of DNA that are complementary to the sequence of the target DNA and can be designed to bind to the pathogen’s genetic material with high specificity. The polymerase is the protein that catalyzes the addition of dNTPs, the building blocks that make up DNA sequences, to the primer. With the buffer keeping pH and salt concentrations at an ideal level for polymerase activity, the polymerase can effectively elongate the primers using the target DNA strand as a template, and eventually create a complementary strand.
The PCR technique shown in Figure 2 uses alternating heating and cooling cycles consisting of three phases: (1) denaturation of the DNA strands, (2) the binding of the DNA primers to the template, and (3) the creation of the complementary strand. During these steps, double-stranded DNA separates and each strand is replicated; this process is repeated again and again with each heating and cooling cycle. Eventually, billions of copies of the pathogen’s DNA are produced.
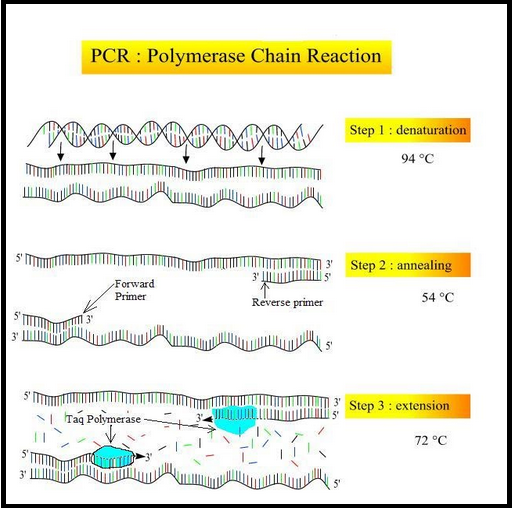
During the first phase, a sample containing a pathogen’s double-stranded DNA is heated in a thermocycler to ~95°C. At this temperature, the hydrogen bonds that link the two strands are disrupted, and the strands separate entirely. This temperature, however, is far too high to allow for primers to effectively bind, or anneal, to the separated DNA strands. So, the second phase of PCR is initiated: the thermocycler is cooled to ~50°C to allow the primers to anneal to its complement on the template DNA’s top and bottom strands. Once the primers have annealed, the third stage is initiated. The sample is heated again to 72°C, as this is the optimal temperature for the polymerase to extend the bound primers by adding dNTPs that are complementary to the template strands. At the end of this stage, two strands complementary to the original DNA template have been synthesized; therefore, the result of this single, 5-minute heating and cooling cycle is an effective doubling of the original DNA content. After repeating this process 30 times or more, the power of PCR is fully apparent: more than a billion copies can be produced from a single DNA piece in less than an hour, allowing for effective pathogen detection and analysis. If the sample contains no virus, the primer has no template to bind to, and the result is little to no amplification.
In the case of pathogens with an RNA genome, such as viruses in the coronavirus family, an additional step of converting the RNA into DNA is required prior to the PCR reaction. This is accomplished using reverse transcription, illustrated in Figure 3 below. The RNA genome of the virus can be used as a template to produce its more stable, complementary DNA (cDNA) counterpart using the reverse transcriptase protein. Once this cDNA is produced, it can be used in the traditional PCR workflow. This process is called “reverse” transcription because it is the opposite of what typically happens in human cells, in which RNA is always produced from the DNA template. Typically, around a thousand copies of the virus’ RNA or DNA product are required for a successful PCR reaction.
Not only can patient samples yield a simple positive or negative result for viral presence, but quantification of the amount of virus product in a sample can also be determined using qPCR (quantitative polymerase chain reaction). This can be detected in two ways at specific points during the qPCR reaction: (1) general fluorescent reporters that bind to double-stranded DNA, or (2) fluorescently labeled DNA fragments, also known as DNA probes, that emit light once it binds to a specific DNA sequence. This fluorescence is detected and measured by a real-time PCR instrument.

The number of amplification cycles required for a fluorescent signal to be detected is called the cycle threshold (CT) value. CT value and DNA content have an inverse relationship - the higher the CT value, the lower the DNA content in the original sample. By comparing the CT values of a patient’s sample to that of a set of virus standards with known copy numbers, the approximate number of DNA or RNA viral copies in the original sample can be defined. This is useful for clinicians assessing their patients’ prognosis and their risk of transmitting the virus to others.
However, our interactions with the world are often far too messy and frequent to guarantee infection by only one virus when we’re sick; it is possible for us to be co-infected with different viral pathogens at the same time. qPCR protocols can be tweaked to resolve this challenge by utilizing sequence-specific probes with different fluorescent signals that bind to multiple types of viruses in a single reaction. Real-time PCR instruments can independently analyze each fluorescent signal, thus confirming the presence and quantifying the viral load of each pathogen in the patient’s sample.
Novel pathogens, sadly enough, will continue to emerge in the near future. Thus, designing qPCR tests with accessibility and efficiency in mind is paramount for successful medical and public health responses. We need to invent instruments for fast and convenient detection using easily deployable point-of-care systems. Now let’s get to the engineering required to make this happen.
At SN&N Electronics, we have designed and engineered several thermal cycling systems for DNA/RNA amplification. The sample (COVID-19 detection requires a swab from the nose and throat) is deposited into a small well in a disposable cartridge that also has several chambers containing primers, polymerase, buffers and other reagents required by the PCR process. The key PCR idea is to rapidly heat and cool the sample that contains a miniscule amount of DNA to specific temperatures and hold the temperatures within a precise band for a specified time period as shown in Figure 4.
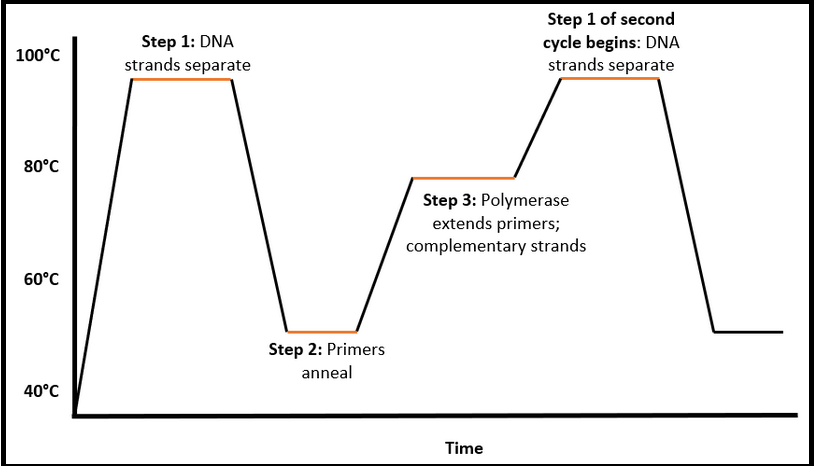
Figure 4: Thermal process of one PCR cycle. Cycle durations are a few minutes, ramp-up and ramp-down periods a few seconds and cycle count could be anywhere between ten and one hundred. (Image © SNN)
This results in the exponential growth of DNA for subsequent or concurrent detection using fluorescence techniques described earlier or other methods such as counting nanoparticles that adhere to the amplified DNA. In order to speed up the amplification process and consequently reduce the total analysis time, it is good to minimize the temperature ramp-up and ramp-down time periods. In SN&N designs, we have achieved rapid heating rates of greater than 20°C per second accomplished using a resistive heater or a bipolar thermoelectric cooler (TEC) operated in the “heat” mode. The electronics that drive the heater are carefully tuned to ensure there is minimal temperature overshoot during the rapid heating stage. These special tuning methods are, in the language of electrical engineering, negative feedback PID (proportional- integral-derivative) controllers. These techniques ensure the thermal response is fast, with minimal overshoot, and with a rapid settling time with negligible fluctuations at the hold temperature in the chamber of the cartridge that holds the sample. The SN&N thermal controllers ensure the target temperature matches the setpoint temperature to within 0.1°C. Cooling can be accomplished using a fan or the TEC operated in “cool” mode to allow even faster cooling rates. A heater assembly and a disposable cartridge containing the sample, primers, polymerase, buffers etc. undergoing PCR cycling in the Integrated Nano-Technologies Palladium instrument is shown in Figure 5.

Figure 5: The heater assembly for PCR and the syringe motor used for sample-prep is shown on left. On right, the cartridge containing the sample is fully engaged and going through the PCR process. (Courtesy: Integrated Nano-Technologies and CITO Medical)
Often, the DNA/RNA is lodged in a cell and needs to be broken out before PCR amplification using a process called cell lysing. Cell lysing is often accomplished by the delivery of ultrasonic energy to the cells to literally “shake-out” the DNA. SN&N Electronics has designed several ultrasonic transducer systems to facilitate cell lysing in an efficient, reliable and repeatable manner. The ultrasonic lysing system requires the design of an electronic drive circuit that efficiently couples high frequency energy to the transducer for conversion to mechanical displacement. The key idea is to operate the transducer in a feedback control loop exactly at its mechanical resonance frequency with the optimum power to provide “vigorous shake and rub” to the cells to crack open the DNA/RNA.

Figure 6: A fully automated DNA/RNA laboratory optimized for point-of-care deployment to enable fast pathogen detection. (Courtesy: Integrated Nano Technologies)
Additionally, before amplification, the sample needs to be rapidly spun to remove extraneous cellular debris, filtered, and have various primers, polymerase and reagents added and mixed before it is ready for the PCR process. This preliminary step is called “sample-prep” and is accomplished by tiny but powerful electric motors that function as syringes to add and mix precise volumes of primers, polymerase, reagents etc. into the sample for facilitating the amplification process as shown in Figure 2. SN&N has designed sample-prep systems that are completely automated and work in conjunction with ultrasonic lysing, PCR amplification and final detection based on fluorescence or a nanoparticle count to provide the positive/negative test result for a pathogen or other biological matter. This automated laboratory is operated by well-crafted real-time code residing in a set of microprocessors and controlled by top-level software in a laptop, tablet or amazingly enough, a phone connected to the internet! A picture of a battery operated “DNA laboratory” controlled by a phone with the footprint of a small book is shown in Figure 6 and is at the cutting edge of modern, efficient and fast pathogen detection systems.
We invite interested parties to talk to us about their molecular diagnostics ideas for which SN&N Electronics can provide a turnkey solution that integrates all aspects of sample-prep, ultrasonic cell lysing, PCR, optical or nanoparticles based detection, connection to the cloud, database management etc. in an automated “put-in-the-cartridge, go-for-a-coffee and get-the-result” system! As you can see, clean modern aesthetics are an integral part of the design besides wonderfully crafted functionality. Come work with us on solutions to the most compelling and urgent task today, pathogen detection!